
- La modélisation des matériaux à l’échelle atomique donne accès à trois types d’informations : a) la description de la structure électronique pour un ensemble donné de coordonnées atomiques, b) l’optimisation de la structure atomique sous l’action des forces dérivée directement de la structure électronique et c) l’évolution temporelle des coordonnées atomiques qui conduit à la prise en compte des effets liés à la température. Cette évolution a lieu sous l’action des forces qui sont des fonctions explicites des degrés de liberté électroniques. Ces trois ingrédients sont combinés dans un schéma de calcul auto-cohérent, la dynamique moléculaire « ab-initio » (DMAI).
- La DMAI est l’outil de choix pour obtenir les propriétés structurales, électroniques et dynamiques à température finie, par le biais des moyennes temporelles (i.e. les moyennes statistiques dans la limite ergodique) sans faire recours à des paramètres ajustables extraits des données expérimentales. L’approche de la DMAI est le dénominateur commun des activités de modélisation à l’échelle atomique actuellement poursuivies au sein du DCMI.La DMAI a une double origine conceptuelle. Du côté de la structure électronique, la DMAI est basée sur la théorie de la fonctionnelle de la densité (DFT) qui donne accès aux propriétés dépendant de la liaison chimique. Tous les électrons de valence présents dans le système (qui en principe est une collection de n corps) sont traités d’une manière globale comme une distribution de densité qui s’exprime sous la forme :
{kind=link}
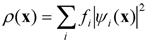
où les ψi(x) sont des fonctions d’onde à un électron et fi les nombres d’occupation ( fi = 1 pour chaque orbitale occupée). Ce cadre est l’ingrédient clé pour obtenir des forces interatomiques fiables, donnant accès à l’évolution temporelle à partir d’une configuration donnée, qui a lieu en suivant les lois de la mécanique statistique. La DMAI établit le lien entre les variables microscopiques (les degrés de liberté atomiques et électroniques) et les propriétés macroscopiques accessibles expérimentalement. La DMAI ouvre aussi la voie à l’observation directe des mécanismes régissant le mouvement atomique et, plus généralement, à un ensemble d’informations non accessibles aux expériences.Nous utilisons deux versions de la DMAI. Dans la première (méthode dite de Car-Parrinello) les orbitales électroniques sont traitées comme une variable dynamique qui suit d’une manière adiabatique et auto-cohérente le mouvement des ions. Dans la deuxième, (méthode dite de Born-Oppenheimer), la structure électronique est optimisée pour chaque configuration atomique pendant l’évolution temporelle. Pour garantir le couplage adiabatique, la méthode Car-Parrinello est mieux adaptée aux systèmes ayant un gap finie dans la densité d’état électronique. Par voie de conséquence, l’utilisation de l’approche Born-Oppenheimer est conseillée lorsque l’on s’intéresse aux métaux (voir dans la suite pour une approche alternative qui permet de traiter ces cas spécifiques).L’application de la DMAI dans sa version standard ne permet pas de décrire des processus activés qui se déroulent sur des échelles temporelles bien au delà de celles couramment accessibles aux architectures de calcul les plus performantes (à titre d’exemple, 100-500 ps au plus pour des systèmes composés de 100-500 atomes). Pour ces raisons, la DMAI peut (et doit) être utilisée en combinaison avec des schémas conçus pour accélérer l’échantillonnage dans l’espace des configurations. Au sein de notre équipe nous employons les méthodes dites de la métadynamique and le « blue moon » pour explorer le profil de valeurs de l’énergie libre. Ceci s’effectue par le biais d’un ensemble de coordonnées de réaction, choisies de manière à décrire les degrés de liberté qui varient lentement dans l’espace. Cette démarche consiste à suivre l’évolution dynamique de réactions qui mettent en jeu des mécanismes de migration et/ou des réactions chimiques, dans le cadre d’une extension de la DMAI que l’on peut définir DMAI « réactive ».Les excitations électroniques caractérisées par des valeurs bien définies de la température électroniques peuvent être décrites par une extension de la DMAI qui s’appuie sur l’idée d’énergie libre (EL-DMAI) pour tenir compte des occupations électroniques dépendantes de la température. Dans ce cas les degrés de liberté électroniques ne sont pas maintenus à l’etat fondamental sur la surface de Born-Oppenheimer et ils n’évoluent pas de manière adiabatique (comme dans la méthode Car-Parrinello). Au contraire, ils se voient attribuer une température donnée, qui correspond à une distribution de Fermi-Dirac des occupations fi associées à chaque orbitale. Plus spécifiquement, l’équation (1) reste valable mais les occupations des orbitales prennent la forme
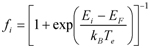
où Ei est l’énergie de la valeur propre de l’orbitale ψi(x), EF l’énergie du niveau de Fermi du système, kB la constante de Boltzmann et Te la température correspondant à l’excitation électronique. Dans ce schéma, la structure ionique évolue d’une manière auto-cohérente sous l’action des forces calculées à partir de la fonctionnelle EL-DMAI. Ces mêmes coordonnées changent aussi en fonction des excitations électroniques.Une stratégie qui comporte, en fonction du positionnement dans l’espace des atomes, des degrés de précision différents dans la description des forces, est très avantageuse et techniquement à la portée de la simulation à l’ordinateur. Il s’agit d’identifier un sous-ensemble restreint d’atomes et d’étudier en détail leur comportement vis-à-vis de la liaison chimique. Ce sous-ensemble est traité par la DMAI. La partie restante du système fait l’objet d’une description des interactions à l’aide des potentiels effectifs d’interaction, qui ne dépendent pas explicitement de la structure électronique. Cette approche est connue sous le nom de QM/MM (en anglais, quantum mechanics molecular mechanics). Au sein du DCMI la méthode QM/MM est utilisée pour étudier des ensembles moléculaires étendus et les réactions chimiques qui ont lieu à l’intérieur de ces structures.
Contacts :